No cross, no crown.
不经历风雨,怎么见彩虹
Updated time: 01/19 2024.
Ref
Theory and implementation behind: Universal surface creation - smallest unitcell Bjarke Brink Buus, Jakob Howalt & Thomas Bligaard August 2, 2019
vaspkit得到标准化原胞基矢和pymatgen得到标准化原胞基矢差异 - VASPKIT使用相关 - VASPKIT论坛 - VASPKIT
写在前面的话
我复现的是关于薄膜的单轴,双轴拉伸。本来想基于 ASE 的 Atoms 创建一个子类 Films,但是我不太能理清其中的继承关系,做到一半发现有部分函数调用失败,于是还是归为 Atoms 类。
1. ASE 中的 surface
参考 [1],ASE 中的 surface 包括 common surface 和 non-common surface。为了使得理解的更为透彻,也方便后面的修改,这里我主要了解的是 non-common surface 的理论介绍和源代码。
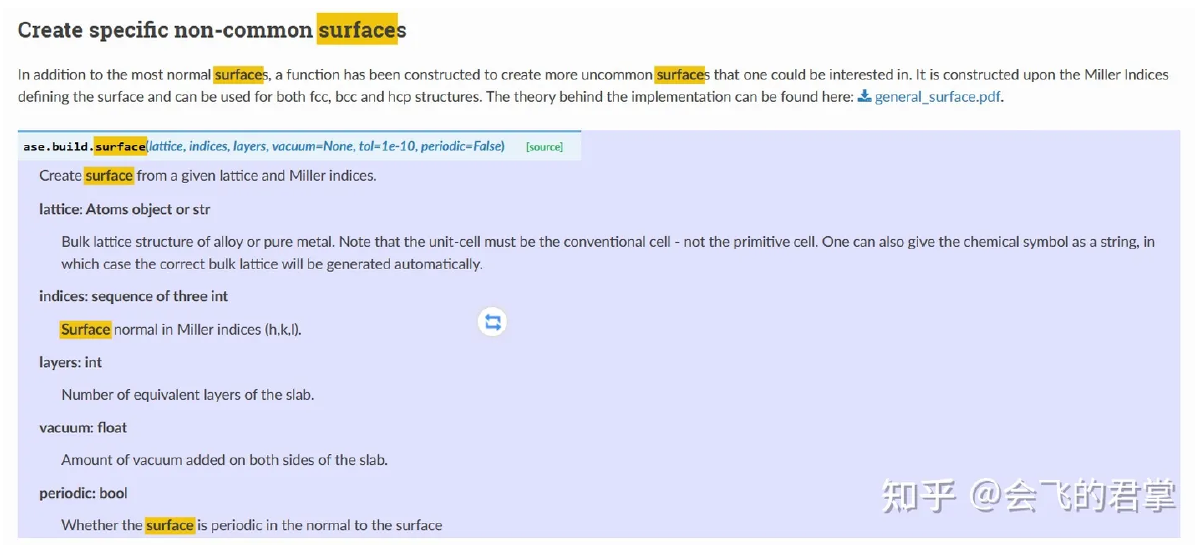
对于参数的理解有,lattice 不能是原胞,必须是 conventional cell; indices 的单位是 (a1, a2, a3), 而不是 (x,y,z); periodic 只决定 z 方向, xy 自动设为 True; layers 是指 layer of slab。在我的修改中,我想指定金属密排面的层数,以及生成的 film 是原胞。
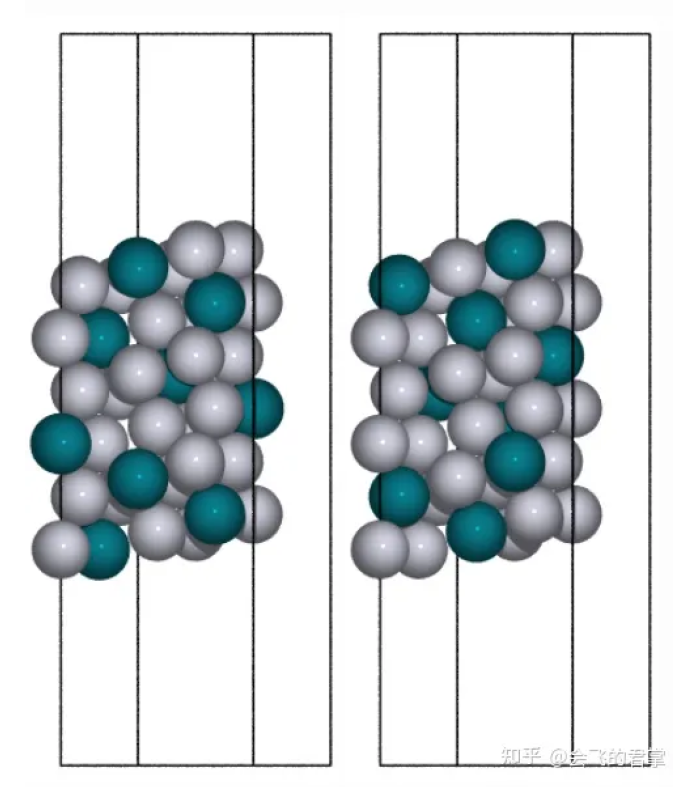
2. 一个简单的例子 (Ref [1])
可以看到这个方法的强大之处在于,我们可以任意指定一个 conventional cell,就可以创建出任意晶向的切面。
从这个例子可以得到:
s3.center(vacuum=10, axis=2)
设定的真空层厚度是可多次变化的,不会累积。
以及源代码中应该考虑到 surface 必然是要加真空层的,所以如果 vacuum = 0, 那么可以看到上下表面的原子紧挨着上下边界。如果 replicate z, 就会发现原子重叠了。
a = 4.0
Pt3Rh = Atoms('Pt3Rh',
scaled_positions=[(0, 0, 0),
(0.5, 0.5, 0),
(0.5, 0, 0.5),
(0, 0.5, 0.5)],
cell=[a, a, a],
pbc=True)
s3 = surface(Pt3Rh, (2, 1, 1), 9)
s3.center(vacuum=10, axis=2)
Pt3Rh.set_chemical_symbols('PtRhPt2')
s4 = surface(Pt3Rh, (2, 1, 1), 9)
s4.center(vacuum=10, axis=2)
3. manual 中的 理论 [ref 3]
3.1 实空间描述
晶面指数 (h k l),与 (a1, a2, a3)轴交叉于 (1/h·a1, 1/k·a2, 1/l·a3)
3.2 倒空间描述
晶面法向量 n = h·b1 + k·b2 + l·b3

3.3 确定面内基矢量
两个基矢量的叉乘应最小,基于此去求解


3.4 确定 v3

3.5 代码中的实现


4 Spglib 寻找原胞 (ref 2)
ref [2] 中有关于寻找原胞的逻辑,先寻找对称性,然后进行转换/旋转操作。
import spglib
lattice = [[5.0759761474456697, 5.0759761474456697, 0],
[-2.8280307701821314, 2.8280307701821314, 0],
[0, 0, 8.57154746]]
points = [[0.0, 0.84688439, 0.1203133],
[0.0, 0.65311561, 0.6203133],
[0.0, 0.34688439, 0.3796867],
[0.0, 0.15311561, 0.8796867],
[0.5, 0.34688439, 0.1203133],
[0.5, 0.15311561, 0.6203133],
[0.5, 0.84688439, 0.3796867],
[0.5, 0.65311561, 0.8796867]]
numbers = [8,] * len(points)
cell = (lattice, points, numbers)
primitive_cell = spglib.standardize_cell(cell, to_primitive=1, no_idealize=1)
print(primitive_cell[0])
However usingstandardize_cell, distortion is not removed for the distorted crystal structure.
由此可见,需要从 Atom 中按照格式输入到函数中。
import spglib
# find primitive cell using spglib
def my_find_prim(atoms: Atoms = None) -> Atoms:
# Convert to a format suitable for spglib
lattice = array(atoms.get_cell())
points = array(atoms.get_scaled_positions())
numbers = array(atoms.get_atomic_numbers())
pbc = array(atoms.get_pbc())
cell = (lattice, points, numbers)
primitive_cell = spglib.standardize_cell(cell, to_primitive=1, no_idealize=1)
# print(primitive_cell)
# primitive_symbols = [chemical_symbols[number] for number in primitive_cell[2]]
# Convert the spglib output back to an ASE Atoms object
primitive_atoms = Atoms(numbers = primitive_cell[2],
scaled_positions = primitive_cell[1],
cell = primitive_cell[0],
pbc=pbc)
return primitive_atoms
5. 设定密排面层数
我的想法是我先生成 1 layer_slab,通过寻找原胞得到这一层slab中有多少原子 num_per_slab,然后除以(预先设定的)一层密排面中有多少高度不一致的原子 number_per_layer(例如 FCC,HCP 都为1,二硫化钼为3),得到该创建几层slab,num_rep_z
# find number of atoms per slab
def my_find_num_per_slab(my_bulk: Atoms = None,
slice_plane: tuple[int] = (0, 0, 1),
my_tol: float = 1e-6,
my_periodic: bool = False,
number_per_layer: float = None) -> float:
my_one_slab = surface(my_bulk, slice_plane , 1, vacuum = 20, tol=my_tol, periodic=my_periodic)
prim_one_slab = my_find_prim(my_one_slab)
atom_number_per_slab = len(prim_one_slab.get_positions())
layer_number_per_slab = atom_number_per_slab/number_per_layer
return layer_number_per_slab
主函数
from numpy import sqrt, ndarray, array, int32
from os import makedirs, path
from inspect import stack
from ase.cell import Cell
from ase.symbols import Symbols
import numpy as np
from ase import Atoms
from ase.data import chemical_symbols
from ase.build import bulk, surface
from ase.io import write
from ase.visualize import view
from ase.io.vasp import _symbol_count_from_symbols, _write_symbol_count
from ase.utils import reader, writer
import re
from pathlib import Path
from typing import List, Optional, TextIO, Tuple
import spglib
def generate_film( symbols: str = None, # str
structure: str = None, # str
num_layers: int = None, # int
replic_z: int = None, # int
my_vacuum: float = None, # float
slice_plane: tuple[int] = (0, 0, 1), # Tuple of ints
my_tol: float = 1e-6, # float
my_periodic: bool = False, # bool
a_fcc: float = 2.95*sqrt(2.0), # float
a_hcp: float = 2.95, # float
my_covera: float = sqrt(8.0/3.0), # float
move_atom: list = [0.1, 0.1, 0.0],
number_per_layer: float = 1.0
) :
# parameters : 1-8 line: general setting
# 9 10-11 line: fcc, hcp parameters
# when we use surface() function, my_bulk must be a conventional cell, not a primitive cell, so set cubic=True
if structure == 'fcc':
my_bulk = bulk(symbols, structure, a=a_fcc, cubic=True)
elif structure == 'hcp':
my_bulk = bulk(symbols, structure, a = a_hcp, covera = my_covera, cubic=True)
else:
raise ValueError('%s is an invalid structure' % structure)
layer_number_per_slab = my_find_num_per_slab(my_bulk, slice_plane, my_tol, my_periodic, number_per_layer)
# print('layer_number_per_slab: %s' % layer_number_per_slab)
if num_layers:
num_rep_z = int(num_layers/layer_number_per_slab)
elif replic_z:
num_rep_z = replic_z
else:
raise ValueError('%s or %s is an invalid value' % num_layers % num_rep_z)
# print('rep_z: %s' %num_rep_z)
my_slab = surface(my_bulk, slice_plane , num_rep_z, vacuum = my_vacuum, tol=my_tol, periodic=my_periodic)
my_slab = my_find_prim(my_slab)
move_atoms(my_slab, move_atom)
my_slab.wrap()
return my_slab
# move atoms
def move_atoms(atoms: Atoms = None,
translate_matrix: ndarray = array([0.1, 0.1, 0.0])) -> Atoms :
scaled = atoms.get_scaled_positions()
scaled += translate_matrix
atoms.set_scaled_positions(scaled)
atoms.wrap()
return atoms
# find primitive cell using spglib
def my_find_prim(atoms: Atoms = None) -> Atoms:
# Convert to a format suitable for spglib
lattice = array(atoms.get_cell())
points = array(atoms.get_scaled_positions())
numbers = array(atoms.get_atomic_numbers())
pbc = array(atoms.get_pbc())
cell = (lattice, points, numbers)
primitive_cell = spglib.standardize_cell(cell, to_primitive=1, no_idealize=1)
# Convert the spglib output back to an ASE Atoms object
primitive_atoms = Atoms(numbers = primitive_cell[2],
scaled_positions = primitive_cell[1],
cell = primitive_cell[0],
pbc=pbc)
return primitive_atoms
至此,我想要的功能都实现了。
写在最后的话
未解决的问题
- 我其实搞不懂这个surface函数究竟是怎么转出来的,当我没有添加得到原胞的功能时,FCC {111} 1 层slab里有一层密排面,4个原子。FCC {100} 1 层slab里有两层原子面,4个原子。
film = generate_film(symbols = 'Au', structure = 'fcc', num_layers = 1, my_vacuum = 20, slice_plane = (1 0 0) or (1,1,1), a_fcc = 2.95*sqrt(2.0))
正是因为没搞懂,所以为了实现设定原子密排面数量,我采用了上述转换。
Please indicate the source when reprinting. Please verify the citation sources in the article and point out any errors or unclear expressions.